Die Hämophagozytische Lymphohistiozytose (HLH), englische Synonyme hemophagocytic syndrome (HPS), reactive hemophagocytic syndrome (RHS), macrophage activation syndrome (MAS, deutsch Makrophagenaktivierungssyndrom) oder lymphohistiocytic syndrome (LHS), ist eine seltene, außerordentlich schwer verlaufende hyperinflammatorische [1] Erkrankung des Immunsystems, welche durch hohes Fieber
Hemophagocytic lymphohistiocytosis (HLH) is a hyperferritinemic hyperinflammatory syndrome. A primary hereditary form can be distinguished from a secondary acquired form. In adults the secondary form accounts for the vast majority of cases. Die HLH ist eine potenziell lebensbedrohliche Erkrankung und ein relevanter Teil der betroffenen
Die (primäre) erworbene Hämophagozytische Lymphohistiozytose (HLH) ist ein den Histiozytosen zugeordnetes hyperinflammatorisches Syndrom, verursacht durch eine massive, überschießende, sepsisartige, inflammatorische Reaktion des Immunsystems mit überwältigender Aktivierung von T-Lymphozyten und Makrophagen. Die Erkrankung ist lebensbedrohlich.
The risk of children developing treatment-related AML in the HLH-2004 and HLH-94 studies was 0.3% (1/368) to 0.4% (1/249) at a median follow-up of 5.2 and 6.2 years, respectively, 3,5,6 and 1.2% (1/81) in patients with EBV-associated HLH (EBV-HLH) treated with a median cumulative etoposide dose of 1500 mg/m 2 body surface area, with a median follow-up of 44 months. 60 The need to stay below a
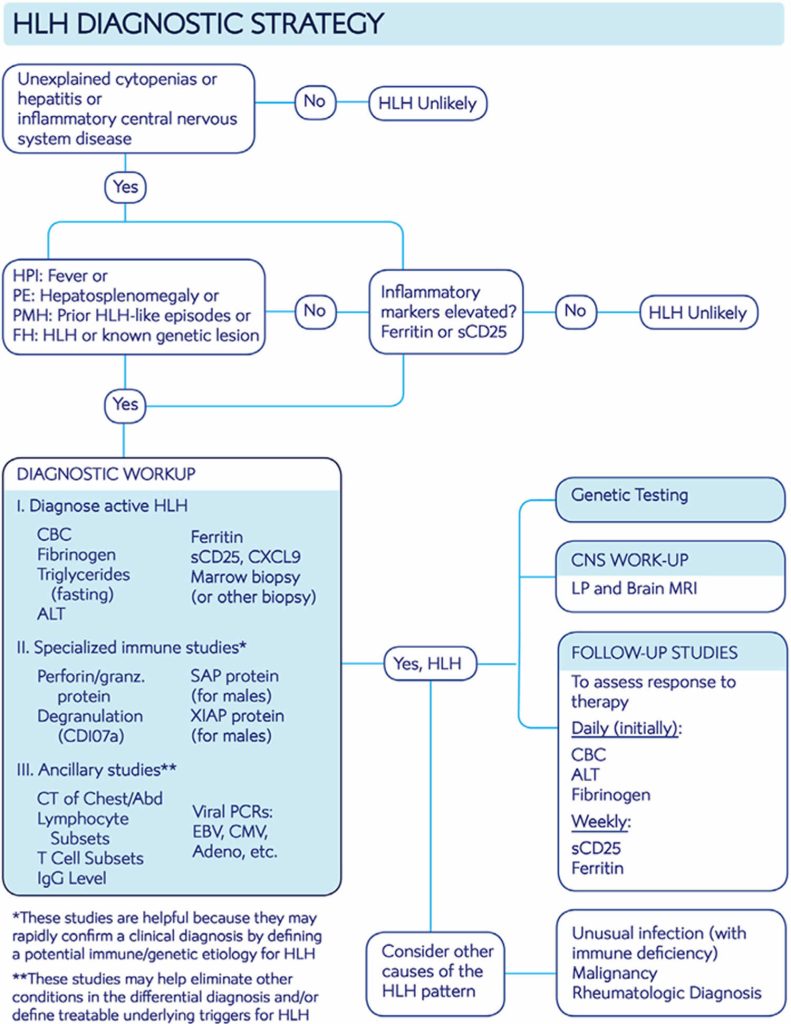
Molecular diagnosis consistent with HLH: pathologic mutations of PRF1,UNC13D, Munc18-2, Rab27a, STX11, SH2D1A, or BIRC4. or. Five of these eight criteria: Fever; Splenomegaly; Cytopenia of two or more cell lines (i.e. Hb <90 g/L, platelets <100 x 10E9/L, neutrophils <1 x 10E3/L) Either elevated triglycerides or low fibrinogen
Hemophagocytic lymphohistiocytosis (HLH) is a hyperferritinemic hyperinflammatory syndrome. A primary hereditary form can be distinguished from a secondary acquired form. In adults the secondary form accounts for the vast majority of cases. Die HLH ist eine potenziell lebensbedrohliche Erkrankung und ein relevanter Teil der betroffenen Die (primäre) erworbene Hämophagozytische Lymphohistiozytose (HLH) ist ein den Histiozytosen zugeordnetes hyperinflammatorisches Syndrom, verursacht durch eine massive, überschießende, sepsisartige, inflammatorische Reaktion des Immunsystems mit überwältigender Aktivierung von T-Lymphozyten und Makrophagen. Die Erkrankung ist lebensbedrohlich.
The risk of children developing treatment-related AML in the HLH-2004 and HLH-94 studies was 0.3% (1/368) to 0.4% (1/249) at a median follow-up of 5.2 and 6.2 years, respectively, 3,5,6 and 1.2% (1/81) in patients with EBV-associated HLH (EBV-HLH) treated with a median cumulative etoposide dose of 1500 mg/m 2 body surface area, with a median follow-up of 44 months. 60 The need to stay below a Molecular diagnosis consistent with HLH: pathologic mutations of PRF1,UNC13D, Munc18-2, Rab27a, STX11, SH2D1A, or BIRC4. or. Five of these eight criteria: Fever; Splenomegaly; Cytopenia of two or more cell lines (i.e. Hb <90 g/L, platelets <100 x 10E9/L, neutrophils <1 x 10E3/L) Either elevated triglycerides or low fibrinogen
The possible role of infectious diseases in causing HLH was first elucidated in 1979 in a case series describing renal transplant patients with a viral-associated HLH (viral-associated hemophagocytic syndrome). 4 EBV, a DNA (deoxyribonucleic acid) virus and member of the Herpesviridae family has been the most consistently reported virus associated with HLH. 36 - 52 The majority of EBV
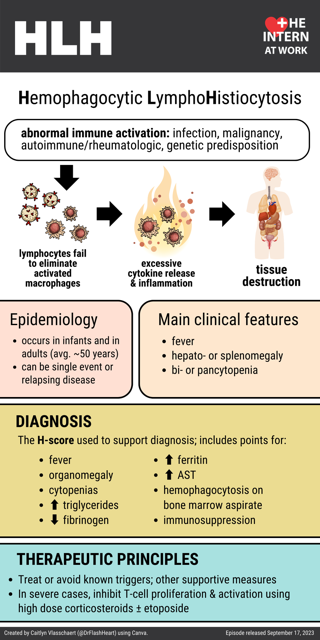
In hematology, hemophagocytic lymphohistiocytosis (HLH), also known as haemophagocytic lymphohistiocytosis (British spelling), and hemophagocytic or haemophagocytic syndrome, [1] is an uncommon hematologic disorder seen more often in children than in adults. It is a life-threatening disease of severe hyperinflammation caused by uncontrolled proliferation of benign lymphocytes and macrophages
Clinical and testing criteria. Hemophagocytic lymphohistiocytosis can be diagnosed if there is a mutation in a known HLH-associated gene or if at least 5 of 8 diagnostic criteria based on HLH-2004 protocol (1, 2) are met: Fever (peak temperature of > 38.5° C for > 7 days). Splenomegaly (spleen palpable > 3 cm below costal margin). Cytopenia involving > 2 cell lines (hemoglobin < 9 g/dL [90 g
Die Prognose der sekundären HLH ist nach der Behandlung der ursächlich auslösenden Erkrankung gut. Auch die sekundäre HLH ist eine ernste Erkrankung, aber schwere Verläufe sind seltener. Es kann zu Rückfällen kommen, wenn die Grunderkrankung (z.B. schweres Rheuma) nicht zur Ruhe kommt, aber eine Stammzelltransplantation ist in der Regel
Hämophagozytose-Syndrom (HLH) - Universitätsklinikum Freiburg
The possible role of infectious diseases in causing HLH was first elucidated in 1979 in a case series describing renal transplant patients with a viral-associated HLH (viral-associated hemophagocytic syndrome). 4 EBV, a DNA (deoxyribonucleic acid) virus and member of the Herpesviridae family has been the most consistently reported virus associated with HLH. 36 - 52 The majority of EBV In hematology, hemophagocytic lymphohistiocytosis (HLH), also known as haemophagocytic lymphohistiocytosis (British spelling), and hemophagocytic or haemophagocytic syndrome, [1] is an uncommon hematologic disorder seen more often in children than in adults. It is a life-threatening disease of severe hyperinflammation caused by uncontrolled proliferation of benign lymphocytes and macrophages Clinical and testing criteria. Hemophagocytic lymphohistiocytosis can be diagnosed if there is a mutation in a known HLH-associated gene or if at least 5 of 8 diagnostic criteria based on HLH-2004 protocol (1, 2) are met: Fever (peak temperature of > 38.5° C for > 7 days).
Splenomegaly (spleen palpable > 3 cm below costal margin). Cytopenia involving > 2 cell lines (hemoglobin < 9 g/dL [90 g Die Prognose der sekundären HLH ist nach der Behandlung der ursächlich auslösenden Erkrankung gut. Auch die sekundäre HLH ist eine ernste Erkrankung, aber schwere Verläufe sind seltener. Es kann zu Rückfällen kommen, wenn die Grunderkrankung (z.B. schweres Rheuma) nicht zur Ruhe kommt, aber eine Stammzelltransplantation ist in der Regel
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammation syndrome. In adults, secondary HLH is mostly observed. HLH is often triggered by infections, malignancies or autoimmune disorders. Daher wird bei Patienten mit COVID-19-Erkrankung, die Sauerstoff benötigen oder invasiv beatmet werden, die Gabe von Dexamethason empfohlen.
Weiterhin möchten wir dafür werben, Patienten in das HLH-Register für Erwachsene einzubringen, um mehr Erkenntnisse über ein Krankheitssyndrom zu gewinnen, das immer noch mit einer hohen Mortalität einhergeht. Patienten und Angehörige sollen allgemeinverständliche Informationen zur HLH erhalten. Beachten Sie bitte auch unseren Flyer
HLH/MAS stellt keine einheitliche Erkrankung dar, sondern vielmehr einen immunpathologischen Zustand, der im Rahmen verschiedener Grunderkrankungen bzw. klinischer Konstellationen auftreten kann und die Maximalausprägung einer systemischen Hyperinflammation darstellt [].Die Pathophysiologie von HLH/MAS ist bisher unzureichend verstanden.
Die hämophagozytische Lymphohistiozytose (HLH) ist eine sehr seltene Erkrankung, die durch einen angeborenen Immundefekt oder eine erworbene Immundysregulation verursacht wird. Dies führt zu einem schweren, hochentzündlichen Krankheitsbild. Infektiöse Erreger (meist Viren) können den Ausbruch oder Rezidive der Erkrankung auslösen.
Hämophagozytische Lymphohistiozytose (HLH)
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammation syndrome. In adults, secondary HLH is mostly observed. HLH is often triggered by infections, malignancies or autoimmune disorders. Daher wird bei Patienten mit COVID-19-Erkrankung, die Sauerstoff benötigen oder invasiv beatmet werden, die Gabe von Dexamethason empfohlen. Weiterhin möchten wir dafür werben, Patienten in das HLH-Register für Erwachsene einzubringen, um mehr Erkenntnisse über ein Krankheitssyndrom zu gewinnen, das immer noch mit einer hohen Mortalität einhergeht.
Patienten und Angehörige sollen allgemeinverständliche Informationen zur HLH erhalten. Beachten Sie bitte auch unseren Flyer HLH/MAS stellt keine einheitliche Erkrankung dar, sondern vielmehr einen immunpathologischen Zustand, der im Rahmen verschiedener Grunderkrankungen bzw. klinischer Konstellationen auftreten kann und die Maximalausprägung einer systemischen Hyperinflammation darstellt [].Die Pathophysiologie von HLH/MAS ist bisher unzureichend verstanden. Die hämophagozytische Lymphohistiozytose (HLH) ist eine sehr seltene Erkrankung, die durch einen angeborenen Immundefekt oder eine erworbene Immundysregulation verursacht wird. Dies führt zu einem schweren, hochentzündlichen Krankheitsbild.
Infektiöse Erreger (meist Viren) können den Ausbruch oder Rezidive der Erkrankung auslösen.
There are 2 types of HLH: familial and acquired. Familial HLH accounts for about 25% of cases and families pass down the condition. If both parents are genetic carriers of HLH, a child has a 25% chance of having the disease, a 25% chance of not having the disease, and a 50% chance of being a carrier. A number of conditions cause acquired HLH.
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome characterized by hyperferritinemia. A differentiation is made between hereditary and acquired forms. Dennoch konnte für Patienten mit schwerer COVID-19-Erkrankung und starker Entzündungsreaktion gezeigt werden, dass eine immunsuppressive,
Hemophagocytic lymphohistiocytosis (HLH), first defined by Scott and Robb-Smith in 1939, is an uncommon and severe immunological disorder that affects both children and adults, with high mortality rates [1,2,3].HLH, characterized by the overactivation of CD8 + cells and macrophages, which induce local and systemic activation of inflammatory cytokines, including interferon-gamma (IFN-γ), tumor
Die Hämophagozytische Lymphohistiozytose (HLH) ist eine lebensbedrohliche Erkrankung. Entscheidend für die Prognose ist die frühzeitige Diagnosestellung. Formal werden die hereditäre und die erworbene HLH unterschieden. Die hereditären Formen dominieren bei Neugeborenen und Säuglingen, können aber auch im Erwachsenenalter auftreten.
Neue Leitlinie „Hämophagozytische Lymphohistiozytose" - Onkopedia
There are 2 types of HLH: familial and acquired. Familial HLH accounts for about 25% of cases and families pass down the condition. If both parents are genetic carriers of HLH, a child has a 25% chance of having the disease, a 25% chance of not having the disease, and a 50% chance of being a carrier. A number of conditions cause acquired HLH. Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome characterized by hyperferritinemia.
A differentiation is made between hereditary and acquired forms. Dennoch konnte für Patienten mit schwerer COVID-19-Erkrankung und starker Entzündungsreaktion gezeigt werden, dass eine immunsuppressive, Hemophagocytic lymphohistiocytosis (HLH), first defined by Scott and Robb-Smith in 1939, is an uncommon and severe immunological disorder that affects both children and adults, with high mortality rates [1,2,3].HLH, characterized by the overactivation of CD8 + cells and macrophages, which induce local and systemic activation of inflammatory cytokines, including interferon-gamma (IFN-γ), tumor Die Hämophagozytische Lymphohistiozytose (HLH) ist eine lebensbedrohliche Erkrankung. Entscheidend für die Prognose ist die frühzeitige Diagnosestellung. Formal werden die hereditäre und die erworbene HLH unterschieden. Die hereditären Formen dominieren bei Neugeborenen und Säuglingen, können aber auch im Erwachsenenalter auftreten.
In Tabelle 2 sind die derzeit verwendeten Diagnosekriterien (HLH Study Group der Histiocyte Society, Henter et al 2007) aufgeführt. Einen Diagnosepfad, welcher der aktuellen Onkopedia-Leitlinie entnommen ist, zeigt Abbildung Familiäre Erkrankung/bekannter genetischer Defekt; oder. 5 von 8 der typischen Kriterien erfüllt: Fieber:
Die Erkrankung tritt in der Regel im frühen Kindesalter auf, kann sich jedoch auch im Erwachsenenalter manifestieren. Die Inzidenz der sekundären HLH ist ebenfalls schwer abzuschätzen. Die Altersspanne ist hier sehr breit und abhängig von der Ursache. Im Alter steigt der Anteil derer, die eine HLH im Rahmen eines Malignoms entwickeln.
Bei der Hämophagozytischen Lymphohistiozytose handelt es sich um eine Krankheit, die das Immunsystem betrifft. Die Erkrankung tritt generell mit einer sehr geringen Häufigkeit auf. Die Hämophagozytische Lymphohistiozytose ist in der Regel durch einen schweren Verlauf gekennzeichnet. Zudem stellt sie eine sogenannte hyperinflammatorische Krankheit dar.
Er soll dazu beitragen, diese Erkrankung, die Möglichkeiten ihrer Behandlung und die Probleme und besonderen Bedürfnisse der betroffenen Patienten besser zu verstehen. Unsere Informationen ersetzen nicht die erforderlichen klärenden Gespräche mit den behandelnden Ärzten und weiteren Mitarbeitern des Behandlungsteams; sie können aber dabei
Hämophagozytose-Syndrom (Hämophagozytische Lymphohistiozytose, HLH)
In Tabelle 2 sind die derzeit verwendeten Diagnosekriterien (HLH Study Group der Histiocyte Society, Henter et al 2007) aufgeführt. Einen Diagnosepfad, welcher der aktuellen Onkopedia-Leitlinie entnommen ist, zeigt Abbildung Familiäre Erkrankung/bekannter genetischer Defekt; oder. 5 von 8 der typischen Kriterien erfüllt: Fieber: Die Erkrankung tritt in der Regel im frühen Kindesalter auf, kann sich jedoch auch im Erwachsenenalter manifestieren. Die Inzidenz der sekundären HLH ist ebenfalls schwer abzuschätzen. Die Altersspanne ist hier sehr breit und abhängig von der Ursache.
Im Alter steigt der Anteil derer, die eine HLH im Rahmen eines Malignoms entwickeln. Bei der Hämophagozytischen Lymphohistiozytose handelt es sich um eine Krankheit, die das Immunsystem betrifft. Die Erkrankung tritt generell mit einer sehr geringen Häufigkeit auf. Die Hämophagozytische Lymphohistiozytose ist in der Regel durch einen schweren Verlauf gekennzeichnet. Zudem stellt sie eine sogenannte hyperinflammatorische Krankheit dar.
Er soll dazu beitragen, diese Erkrankung, die Möglichkeiten ihrer Behandlung und die Probleme und besonderen Bedürfnisse der betroffenen Patienten besser zu verstehen. Unsere Informationen ersetzen nicht die erforderlichen klärenden Gespräche mit den behandelnden Ärzten und weiteren Mitarbeitern des Behandlungsteams; sie können aber dabei
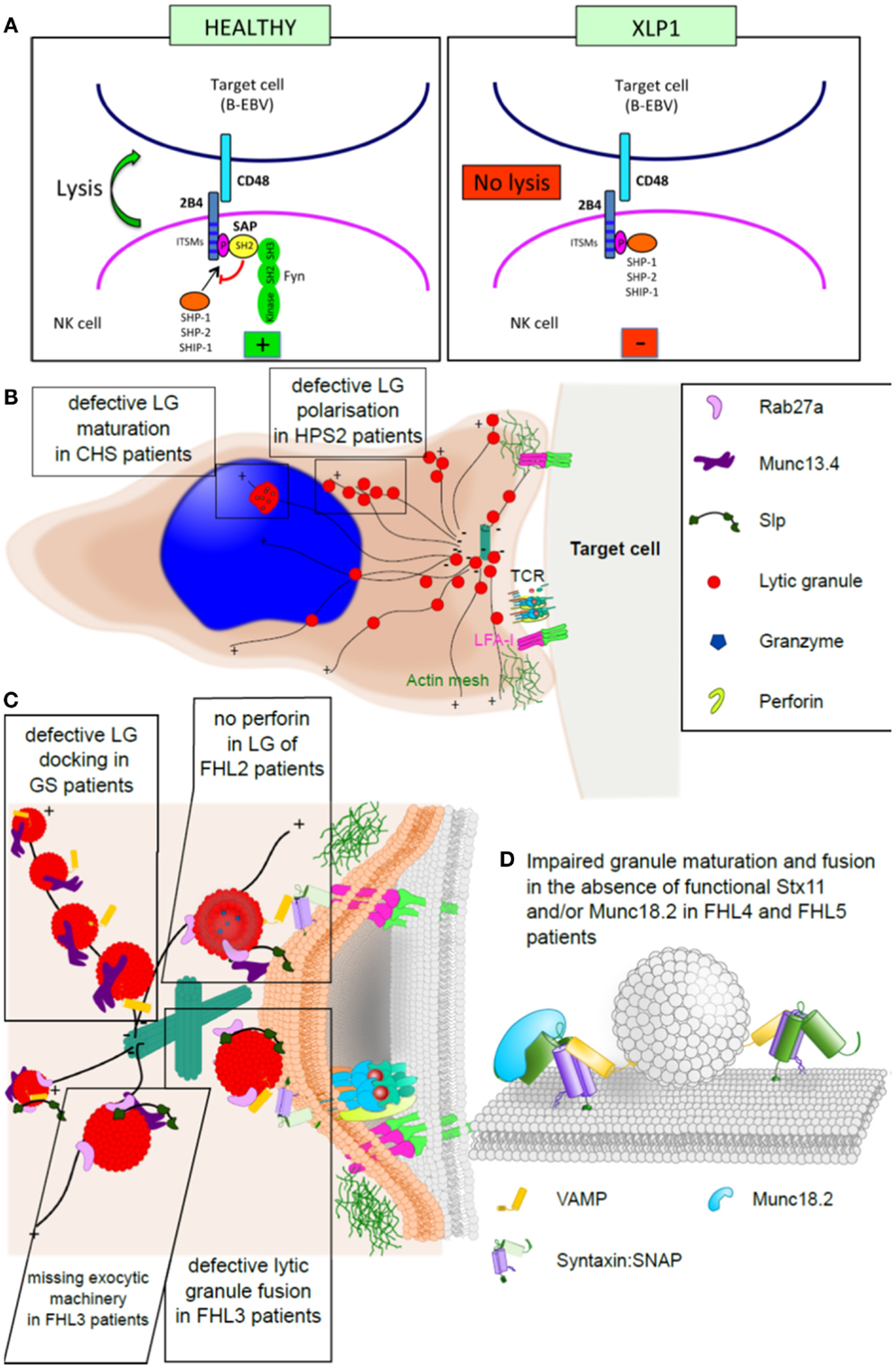
Die HLH ist keine eigenständige Erkrankung. Sie ist gemeinsame Endstrecke eines Immundefekts, welcher genetisch bedingt, oder durch infektiöse, autoimmune, autoinflammatorische, maligne oder auch iatrogene Trigger (Immunsuppression, Stammzelltransplanta-tion) erworben werden kann. Diesem breiten Spektrum
The overall incidence of all forms of HLH was estimated to be 4.2 cases per 1 million population in 2018 in England. 15 The annual incidence of HLH associated with cancer (i.e., malignancy
Die zur Zeit einzige Erfolg versprechende Therapie bei familiärer (hereditärer, primärer) HLH ist die Stammzell-Transplantation. Ohne Stammzellenspender kann die Krankheit nicht geheilt werden.
Wesentlichen zwei Formen der Erkrankung: eine angeborene Form (primäre HLH oder familiäre HLH = FHL) und eine erworbene Form (sekundäre HLH), die nicht angeboren ist. 3.Primäre HLH Bei der primären HLH liegt eine genetische Veranlagung vor, die die Entstehung einer HLH begünstigt.
PDF Hämophagozytose-Syndrom (HLH)
Die HLH ist keine eigenständige Erkrankung. Sie ist gemeinsame Endstrecke eines Immundefekts, welcher genetisch bedingt, oder durch infektiöse, autoimmune, autoinflammatorische, maligne oder auch iatrogene Trigger (Immunsuppression, Stammzelltransplanta-tion) erworben werden kann. Diesem breiten Spektrum The overall incidence of all forms of HLH was estimated to be 4.2 cases per 1 million population in 2018 in England. 15 The annual incidence of HLH associated with cancer (i.e., malignancy Die zur Zeit einzige Erfolg versprechende Therapie bei familiärer (hereditärer, primärer) HLH ist die Stammzell-Transplantation. Ohne Stammzellenspender kann die Krankheit nicht geheilt werden.
Wesentlichen zwei Formen der Erkrankung: eine angeborene Form (primäre HLH oder familiäre HLH = FHL) und eine erworbene Form (sekundäre HLH), die nicht angeboren ist. 3.Primäre HLH Bei der primären HLH liegt eine genetische Veranlagung vor, die die Entstehung einer HLH begünstigt.

.png)